Someone delivers you a little piece of a sophisticated engine and asks you to explain its function in relation to the larger machine. Unless you’re a professional mechanic or an engineering genius, your best bet is to make an informed guess.
This is fundamentally the cell biologist’s predicament. Cryo-electron microscopy (cryo-EM) and the artificial intelligence system AlphaFold provide tools for determining the atomic-scale structure of individual proteins; cutting-edge molecular approaches can reveal where those proteins are active and what they do. However, viewing the real-world interaction of proteins in their native environment — the molecular-scale workings of the cellular engine — remains challenging. However, a new method known as cryo-electron tomography (cryo-ET) is helping to bridge that gap and enabling unparalleled access to the cellular skeleton.
In 2022, structural biologist Peijun Zhang of the University of Oxford in the United Kingdom and her colleagues utilized cryo-ET to decipher the enzymatic pathways that certain bacteria use to absorb and recycle the greenhouse gas methane1. The study revealed how two essential enzymes interact by forming membrane-bound structures that directly funnel one enzyme’s output into the other’s input. “The reaction product is all concentrated in that small compartment so that it would be effective,” Zhang explains. She goes on to say that this discovery could only have been made in the context of entire cells when the complexes remain in their natural state. “You couldn’t do anything like that in vitro.”
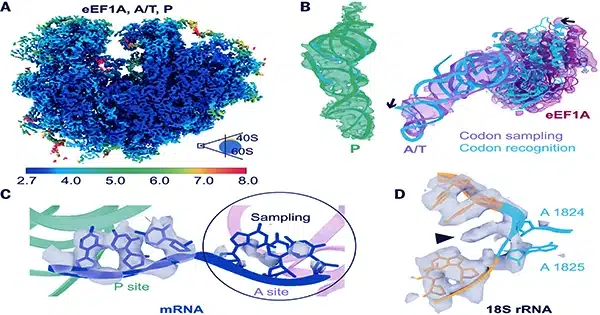
Other researchers are using cryo-ET to learn more about processes including photosynthesis and protein manufacturing, as well as the operation of the nuclear pore complex (NPC), a large protein structure that governs molecular traffic in and out of the nucleus. All of this is part of the developing discipline of visual proteomics, which aspires to describe the biomolecular infrastructure of cells with a resolution comparable to in vitro approaches like cryo-EM. However, cryo-ET is still in its early stages, and data from research that utilizes it frequently challenges clear interpretation.
“Because we see everything, we get to see unexpected things,” says Wolfgang Baumeister, a structural biologist at the Max Planck Institute of Biochemistry in Martinsried, Germany. “But it also comes with the huge challenge of identifying and annotating all of the densities we see in the tomogram.” For the time being, researchers are still figuring out how to distinguish the targets that are most interesting to them in the tightly packed environment of the cell.
Electron microscopy, which dates back to the 1930s, is still a strong method for seeing biological samples at atomic resolution. Purified proteins are flash-frozen into glass-clear ice and scanned in the cryo-EM version. The comprehensive images of these protein molecules, taken at various angles and orientations, are computationally reassembled to produce 3D structures – sometimes with enough precision to discern individual atoms.
Stacking the odds: Cryo-ET involves freezing complete cells rather than isolated proteins and then employing specialized equipment to mill the top and bottom of the sample to create a tiny window known as a lamella. Typically, this is performed by rapidly scanning a focussed ion beam over a target spot to remove extra ice and biological debris. These lamellae are then tilted and photographed from various angles to document the chemical contents before being algorithmically reconstructed into a 3D tomogram and subjected to additional processing and analysis.
The method’s origins may be traced back to Baumeister’s lab, which began researching cryo-ET approximately 40 years ago. He claims that it required decades of effort to get the approach to deliver on its promise. “We could only do what we were hoping to do, which was look at the molecular architecture of cells, less than 20 years ago.” During that time, there have been significant methodological advances, such as the advent of focused-ion-beam milling, ultrasensitive electron detectors, and higher-quality microscopes.
However, none of this makes cryo-ET simple. “It’s not quite structural biology, and it’s not quite cell biology — it’s like some sort of hybrid in between,” explains Elizabeth Villa, a biophysicist at the University of California, San Diego. Unlike cryo-EM, which uses pure protein samples, tomography involves searching for biological structures in their natural context — with a focus on the ‘ search’. “The dirty secret of cryo-ET is that a tomogram covers like 0.001% of a mammalian cell,” Villa explains. As a result, the chances are stacked against any individual lamella possessing the exact protein or event sought by the researcher. With microbial specimens, the possibilities are better — Villa believes that researchers can cover around 30-50% of a bacterial cell with a tomogram — thus microbiology is a significant interest for cryo-ET researchers.
Nonetheless, experts like Villa advise researchers to do their homework before getting started, such as using other ways to learn how abundant a protein is or the elements that influence the frequency with which an event occurs. This is critical since cryo-ET requires hundreds or thousands of samples of the subject of interest to achieve an informative reconstruction.
Even so, research can readily devolve into fishing expeditions. Cell biologist Benjamin Engel of the University of Basel, Switzerland, completed a multi-year effort in 2022 to recreate the cellular machinery used by the algae Chlamydomonas to assemble and maintain its cilia — hair-like structures important in movement and environmental sensing2. “We were just milling these algae cells randomly, trying to hit this structure at the base of the cilium,” Engel explains. “It took many years to gather this one structure.” Faster lamella-generation methods make this endeavor less taxing, and Zhang claims that automation has boosted the number of samples that can be prepared per day from 5 or 6 to 40. Nonetheless, solving a structure can necessitate hundreds of samples.
Shining a cellular spotlight: Researchers can also use correlative light and electron microscopy (CLEM) to limit the cellular terrain that needs to be combed. Prior to freezing, samples are fluorescently labeled to indicate certain proteins or compartments in the cell where an event of interest is most likely to occur. The sample is then imaged using a cryo-fluorescent microscope, allowing researchers to better target lamellae.
Villa and her colleagues, for example, utilized CLEM in 2020 to show how a mutant protein related to Parkinson’s disease interferes with the trafficking of important biomolecules between different areas of the cell3. “Only 30% of the cells even had the phenotype that we were looking for, and if we had just done random lamellae in random places, we would’ve never found it,” Villa adds.
However, as with everything cryo-ET, aligning fluorescence and electron-microscopy data is difficult. Because the resolution of ordinary fluorescence imaging is so much lower than that of electron microscopy, creating ideal lamellae may still necessitate a fortuitous break. “Sometimes your entire image at electron-microscopy magnification would be just one pixel in the light microscope,” University of Oxford structural biologist Kay Grünewald explains. CLEM is being combined with single-molecule-resolution fluorescence microscopy, but this is still a work in progress. For the time being, Grünewald and colleagues frequently utilize tiny fluorescent beads that are electron-dense enough to be observed with an electron microscope, allowing them to more easily triangulate locations between the two imaging systems.
Moving samples between instruments, on the other hand, introduces new challenges, including an increased risk of contamination. And, according to Julia Mahamid, a structural biologist at the European Molecular Biology Laboratory in Heidelberg, Germany, even frozen samples can twist and shift throughout the preparation process. These disturbances may go unnoticed if fluorescence imaging is not used at this point.
These difficulties may be alleviated by a new generation of integrated cryo-CLEM systems. The Chinese Academy of Sciences’ ELI-TriScope4 platform, for example, provides researchers with colocalized electron (E), light (L), and ion (I) beams. One of the project’s primary researchers, structural biologist Fei Sun, says the technology has taken a lot of the guesswork out of his study of the centrioles that sort and split chromosomes during cell division. “Our success rate has increased from around 5% to 90%,” he says. “It’s a huge improvement.”
Meanwhile, because electron microscopy lacks an equivalent of green fluorescent protein, a genetically encoded reporter that can be used for cellular labeling in fluorescence-imaging experiments, researchers are looking for labeling methods that would allow them to zero in on targets of interest without using fluorescence imaging. Existing solutions, such as gold nanoparticles coupled to protein-binding functional groups, are too large and risk artificially aggregating several targets at once. Baumeister is afraid that such tags will disrupt the nanometre-scale features revealed by cryo-ET. Nonetheless, others have observed encouraging outcomes. Grünewald and colleagues, for example, have created DNA-based ‘origami’ tags that fold into electron-dense asymmetrical designs that attach and point to cellular features of interest5.
Although the tags are now limited to exterior proteins, Grünewald is looking at ways to transport them inside the cell. “It’s not as versatile as we’d like it to be yet, but it works quite well for certain questions,” he says.
Combing through the chaos: Following the completion of milling, tilting, and imaging, cryo-ET pictures are reconstructed into a single 3D tomogram, and the real job begins. Scientists must scrutinize each tomogram in order to identify the cellular features of interest.
Even high-quality raw cryo-ET data, however, resembles the grainy monochromatic static of an untuned analog television. Seasoned observers can identify elements such as lipid membranes or mitochondria, but identifying particular proteins or complexes is a more difficult task.
The conventional method is to employ template-matching software, which uses existing structural data to identify the target among the whorls of black and grey. Although rapid, this method is sloppy, and Mahamid points out that even well-studied molecular assemblies like the protein-synthesizing ribosome can sneak through the net. For small, less abundant, or disordered proteins, the method is considerably less dependable — and, by definition, requires prior structural information of the target.
More automation: Deep learning-based structure-prediction algorithms, such as AlphaFold and RoseTTAFold, have shown to be important assets. They enable researchers to make ideas regarding the appearance of poorly understood proteins, as well as reverse-engineer mysterious structures and potentially uncover the protein sequences that formed them. Martin Beck of the Max Planck Institute of Biophysics in Frankfurt, Germany, and his colleagues, for example, used AlphaFold and RoseTTAFold in a comprehensive study of the human NPC6 in June 2022. The final model captured numerous conformations that accounted for more than 90% of the over 1,000 proteins that comprise the NPC, many of which had previously been poorly described.
Deep learning is also assisting researchers in automating the annotation of tomograms. However, some techniques, like the DeePiCt algorithm7 developed by Mahamid and her colleagues, frequently utilize templates. Mahamid describes it as a significant advance over previous template matching methods, but notes that “we’re still missing about 20% of our particles.”
Stefan Raunser’s team at the Max Planck Institute of Molecular Physiology in Dortmund, Germany, described TomoTwin, an alternative technique that can pick and classify particles without prior knowledge of the structure of interest, in April. This strategy, according to Engel, contributes significantly to the goal of automatically constructing detailed protein atlases of a sample. “The one thing I really think we all want is the ‘visual proteomics’ button: press one button and it just gives you everything,” he says.
With enough high-quality particles, cryo-ET can build structures with atomic detail comparable to cryo-EM. This is often accomplished by a procedure known as sub-tomogram averaging, which employs a large number of 3D particle images to build a high-confidence consensus of what the target looks like. Image-processing and refinement methods, such as the Warp and M tools created by computational biologist Dimitry Tegunov at biotechnology business Genentech in South San Francisco, California, can improve the ultimate resolution of these reconstructions. These tools adjust for experimental flaws that degrade the quality of cryo-ET images.
Mahamid used these and other methods in concert with other researchers, including Tegunov, to reconstruct the interaction of a bacterial ribosome with a protein synthesis inhibitor at 3.5-ngström resolution9 — nearly three times the diameter of a hydrogen atom. This was no easy task; Mahamid need 20,000 ribosome examples to construct this model, despite the fact that ribosomes are among the most abundant complexes in the cell.
According to Baumeister, this is a critical constraint. “Probably no more than 10-20% of the proteome is amenable to sub-tomogram averaging,” he estimates. “It requires abundance, and it requires size.” More automation and faster sample processing could help with the former, he says, but smaller and more disordered proteins will likely remain a barrier until raw imaging data quality improves significantly.
Towards Visual Proteomics: Researchers are concerned that as cryo-ET specialists push the technical edge, the field will deteriorate into a race for atomic resolution. “For a lot of the questions that we’re addressing, we don’t need it,” Engel says, adding, “but I wouldn’t turn it down.” Much of Engel’s study, for example, is concerned with the internal machinery that drives photosynthesis in plants, precisely where these structures are located, and how they are arranged. According to him, even a relatively low resolution of 10-15 may be sufficient to handle such concerns.
For the time being, the cryo-ET field is in desperate need of data. Deep learning for image processing and analysis will require a massive archive of annotated images to guide the selection and interpretation of individual particles from the complex biological soup.
More trained employees, as well as access to highly specialized — and expensive — equipment, are also required. According to Baumeister, building a comprehensive cryo-ET procedure can cost up to €10 million (US$10.7 million). “That’s much more than a typical young faculty member’s start-up package,” he says. Indeed, when Mahamid, a former Baumeister lab member, first went looking for teaching positions, her options were constrained by the need for existing cryo-ET capacity. “I could only apply to one place,” she explains.
Nonetheless, momentum is rising, and academics are developing novel applications for the technology. Engel and his colleagues are utilizing portable flash-freezing devices to gather marine specimens from European coastal areas for cryo-ET analysis at their Basel lab. According to Engel, such technology, combined with next-generation computational tools, could one day allow researchers to go from raw samples to molecular inventories, even for totally new species. “If we can push resolution improvements and identify everything using a combination of bioinformatics and imaging, we can really do anything,” Engel says. “It allows for so much more exploration of our world.”